Proteasomes are complex molecular machines that consist of 66 subunits. The assembly of these complexes is highly coordinated in a process that requires at least ten proteasome-specific molecular chaperones. One of the challenges in studying assembly intermediates is their relatively low abundance as compared to the proteasome holoenzyme. Therefore, superior separating techniques are crucial for analyses of proteasomal complexes in general and studies in the assembly in particular. For this reason, native gel analyses have been abundantly used in studying proteasomes, as they provide a high resolution. Native gels are very versatile and can be used in various combinatorial approaches. In this chapter, we outline two approaches to prepare samples for native gels. The first is a yeast cryo-grinding method and the second a core particle (CP) base reconstitution approach. We describe the native gel electrophoresis, as well as various downstream analyses, including 2D native-SDS-PAGE. These techniques and approaches that can all be used, often in parallel, to gain a variety of information about activity and composition of the complexes separated by native gel. The potential combined approaches are highlighted in the summary flow chart in figure 1.
Keywords: Native gel, proteasome, molecular chaperone, assembly, AAA-ATPase, activity assay, LLVY-AMC, fluorescent-label, 2D-gel
1. IntroductionCells contain many large protein complexes that execute a variety of important functions. One well known example is the ribosome, which undergoes a complex process of assembly, maturation, and quality control prior to becoming a functional holoenzyme [1]. While ribosomes are rather unique due to their tight associations of RNA with protein components, they are not alone in their dependence on molecular chaperones to ensure accurate and efficient assembly. Other examples are spliceosomes, nucleosomes, and the proteasomes [2]. Proteasomes are large protease complexes found in the cytosol as well as nucleus of eukaryotic cells. Studying proteasome assembly has provided a wealth of biochemical, cellular, and structural information on the assembly process [3]. Besides key insights into the regulation of this important complex in the cell, it has also revealed several principles that seem to be shared by assembly chaperones in general. Proteasomes consist of a core particle (CP) that is cylindrically shaped as a result of four stacked heptameric rings (α1–7β1–7β1–7α1–7). The proteolytic active sites are localized on the inner surface of this structure. Its formation is guided by proteasome specific chaperones. Half-CP (α1–7β1–6) lacking the β7 subunit is a readily detectable intermediate. The incorporation of β7 coincides with dimerization of two half-CPs, autocatalytic processing of pro-peptides, and the degradation of the chaperone Ump1. This process induces structural changes that lower the affinity for the chaperones Pba1-Pba2 [4–6]. The ends of the CP cylinder serve as docking sites for various regulators of the proteasome, the binding of which are mutually exclusive. These regulators include PA200 (Blm10 in yeast), PI31 (Fub1 in yeast), PA28/REGα, β, γ (all absent from yeast), and the regulatory particle (19S or RP). While the RP is most abundant, the other regulators can be seen on native gels and in purified samples of proteasomal complexes (seeNote 1); for example, Blm10-CP and Blm10-CP-RP can be detected readily in yeast total lysates. The main focus in proteasome research and assembly is on the RP and RP-CP complex for several reasons. First, the RP-CP complexes are the major degradative proteasome complex in the cell. Second, RP contains six AAA-ATPases and is the only known proteasome component to hydrolyze ATP. As such, this complex is responsible for all energy dependent degradation by the proteasome. Third, amongst the regulators only the RP is known to contain a number of ubiquitin receptors and bind ubiquitinated substrates [7]. Biochemical studies have shown that RP can be divided into two subassemblies, base and lid. These complexes have also been identified using native gel analyses of various strains with defects in proteasome assembly. For example, many mutants defective in base assembly accumulate the lid sub-complex [8–11].
One of the challenges in studying assembly is that the immature complexes of interest are a minor species in the cell. The majority of each specific subunit is present in the mature functional complex. Furthermore, it is challenging to distinguish between subcomplexes that are assembly intermediates versus the complexes that result from intracellular or post-lysis dissociation. Nevertheless, there are several approaches available to study this. One approach utilizes unique components that are only present on assembly intermediates. For the proteasome, ten specific assembly chaperones have been identified [12,13]. Five are specific for the RP, known by their yeast names Nas6, Rpn14, Hsm3, Nas2, and Adc17. The other five, Pba1, Pba2, Pba3, Pba4, and Ump1, function in CP assembly. All ten chaperones have orthologues in humans and are specific for proteasome assembly. They have almost exclusively been identified as components of assembly intermediates instead of the holoenzyme. The human orthologue of Nas6, gankyrin, is a notable exception as it is reported to have roles in regulating the degradation of specific factors by the proteasome and is implicated as an oncogene in liver cancer [14]. In general, however, the presence of these chaperones in complexes is considered to be an indicator of the presence of assembly intermediates (seeNote 2). Thus, affinity-tags on these chaperones can be used to purify assembly intermediates [4,9,15]. A second approach is to separate lysates or purified samples based on size or specific biochemical properties, using conditions where mature complexes will behave differently from assembly intermediates. Here, native gels, FPLC, and ultracentrifugation have been commonly used to separate subcomplexes from mature forms [8,9,16]. A third approach to purify intermediates consists of two steps. First, all complexes are purified using an affinity tag. Next, a second tag on a subunit that is only present on the mature complex is utilized to deplete mature complexes from the sample. This approach has been less utilized, at least in proteasome research, due to an initial focus on the chaperone-assisted proteasome assembly and the potential complications resulting from some instability of certain complexes.
Both base and CP rely on assembly chaperones for efficient complex formation whereas lid does not. Lid can assemble without specific chaperones when the subunits are heterologously expressed in bacteria and no lid-specific assembly factors have been identified in yeast or mammals. Recent studies indicate that lid assembly is driven by the subunits themselves [17,18]. These papers also highlight powerful new in vitro approaches to study proteasome assembly, as now the heterologous co-expression of numerous proteasome subunits in E. coli allows for the purification of assembly intermediates that can be used to reconstitute proteasomes.
As described in the preceding sections, the low abundance of assembly intermediates relative to the mature complex can make their analyses challenging. To increase the levels and odds of detecting these assembly intermediates, research in proteasome assembly has heavily relied on genetic manipulations that cause an enrichment in, or disappearance of assembly intermediates. In mammalian cells this has been done using a siRNA approach [19], while in yeast knock-outs and combinations of mutants have been commonly used to manipulate the system [20,8,9]. Consider a conceptually simple example where two subcomplexes A and B combine with other subcomplexes to form a mature complex. Both intermediates can be stable in the cell and accumulate in some small amount. Reducing the abundance of complex B, e.g. by deleting a chaperone associated with complex B, could yield an increase in complex A. This has been observed for the proteasome where the absence of chaperones required for base result in increased levels of lid [9,8,11,10,21]. Lysates or complexes purified from wildtype and genetically manipulated strains, can be analyzed with techniques mentioned above and described below to reveal differences and deduce mechanisms.
Whichever approach has been utilized in studying proteasome assembly, they almost universally utilize native gels as part of the analyses. In the method described here we show a potential flow of how various analyses using native gels at the center can be utilized in your research (Fig. 1).
Figure 1. Flowchart of procedures to analyze proteasome assembly by native PAGE.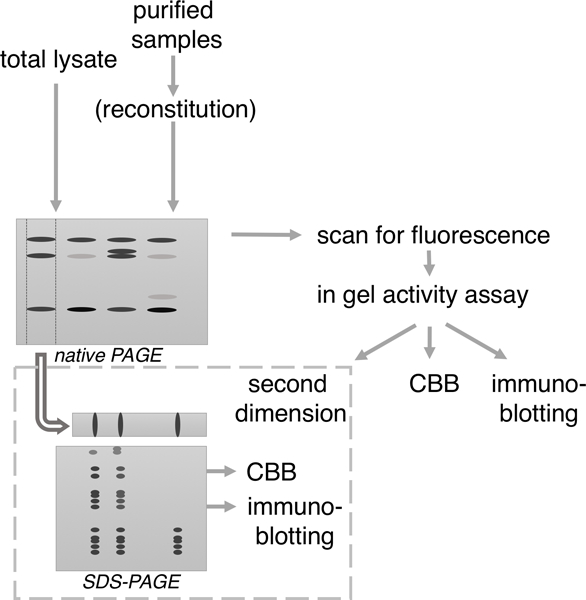
Samples for native gel are generally total lysates or purified proteasome complexes. Following separation, native gels can be analyzed sequentially by scanning for fluorescent proteins (if a subunit was fluorescently-tagged; see figure 2 b left three panels), followed by in gel activity assay (figure 2 b fourth panel). Next gel can be used in one of three ways. Either the gel is stained with Coomassie dye, transferred to PVDF for immunoblotting, or lanes or bands are excised for a second dimension of SDS-PAGE, after which the 2D gel can be stained with Coomassie dye or used for immunoblotting.
2. MaterialsPrepare all solutions using milliQ water (18 mΩ at room temperature, referred to as ddH2O onwards). Procedures are executed at room temperature unless stated otherwise. Standard equipment for protein gel electrophoresis was used for native gels as well as second dimension SDS-PAGE. The same equipment also allows for wet transfer of proteins to PVDF membranes for immunoblotting. We used the mini-PROTEAN electrophoresis system (BIO-RAD) with 1.5 mm thick spacers and combs.
2.1. Total Cell LysateLysis buffer A: 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 1 mM EDTA, 1 mM ATP.
Cryogrinding System (OPS Diagnostics, CG 08–10). This contains porcelain/zirconium mortars, pestles suitable with drill connection, cordless drill, cryocooler with rack.
Liquid nitrogen.
2.2. Purification of BaseLysis buffer B: 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 1 mM EDTA, 1 mM ATP, 10% glycerol.
Wash buffer B: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 1 mM ATP, 10% glycerol.
Dissociation buffer: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM EDTA, 1 M NaCl, 1 mM ATP, 10% glycerol.
Elution buffer B: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.2 mM DTT, 1 mM ATP, 10% glycerol.
IgG resin (MP Biomedicals, 55961).
Cobalt agarose beads.
TEV protease: AcTEV (Invitrogen, 12575–015).
Centrifugal devices for sample concentration (Pall, NanoSEP 10K omega).
2.3. Purification of ChaperonesLysis buffer C: 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 1% Triton X-100 supplemented with protease inhibitors.
Wash buffer C: 1X PBS with 0.1% Triton X-100 and 0.02% sodium azide.
Elution buffer C: 50 mM Tris-HCl (pH6.8), 150 mM NaCl, 1mM EDTA, 1 mM DTT, 0.01% Triton X100.
Glutathione agarose resin.
PreScission Protease (a.k.a C3 protease, GE Healthcare, 27–0843-01).
Protease inhibitor tablets.
Slide-A-Lyzer Dialysis Cassette.
2.4. Reconstitution Assays and Analysis of Chaperone FunctionBuffer A: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM EDTA, 10% glycerol, 1 mM ATP.
Affinity-purified base, lid, and CP.
Affinity-purified chaperones.
ATP regeneration system: 52 mM creatine phosphate, 0.5 mg/ml creatine phosphokinase.
2.5. Native PAGE5x native running buffer: 450 mM Tris-Borate, 25 mM MgCl2 and 2.5 mM EDTA (seeNote 3).
5x native loading buffer: 250 mM Tris-HCl (pH 7.5), 50% glycerol (V/V), ~ 50 ng/ml xylene cyanol.
0.5 M ATP. Dissolve in ddH2O and adjust pH to 7.0 using NaOH. Store as aliquots in −80°C.
40% acrylamide (Acrylamide:Bis Solution 37.5:1).
10% Ammonium persulfate (APS). Dissolve 1 gram APS into 7 ml ddH2O, resuspend and adjust volume to 10 ml to obtain a 10% APS solution (seeNote 4).
TEMED (N,N,N’,N’ – Tetramethylethylene-diamine).
2.6. Detection of Proteasomes in Native Gels2.6.1. Fluorescent Screening of Native GelTyphoon 9410.
2.6.2. LLVY-AMC in Gel Activity AssayStock solutions of 1 M Tris-HCl (pH7.5), 1M MgCl2, and 0.5 M ATP.
Substrate suc-LLVY-AMC (Bachem, I1395) is suspended in dimethylformamide to a concentration of 10 mM, aliquoted and stored at −20°C (seeNotes 5 and 6).
2.6.3. CBB Staining of GelsFixing solution: 50% (V/V) Methanol, 10% (V/V) acetic acid in ddH2O.
Coomassie Brilliant Blue (CBB) G-250 staining solution. Add ~80 mg of Brilliant Blue G in 1 L ddH2O, stir for 2–3 hours using stir bar and plate, add 3 ml 12 N HCl. Keep solution in dark by wrapping bottle in aluminum foil.
Destaining solution: water or 40% (V/V) Methanol, 10% (V/V) Acetic acid in ddH2O.
2.6.4. Immunoblotting of Native GelsMethanol.
Transfer buffer: 25 mM Tris, 190 mM Glycine (optional: 20% methanol).
PVDF membrane.
Whatman paper.
Mini Trans-Blot Module.
2.7. Two Dimensional Native-SDS PAGE.1x SDS sample buffer: 65 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 100 mM DTT 0.01% w/v bromophenol blue.
1x SDS running buffer: 25 mM Tris, 192 mM glycine, 0.1% SDS.
APS, TEMED, and 40% acrylamide (seesection 2.5).
12% SDS-PAGE gel. Mix reagents as indicated in Table 1.
Agarose.
Water-saturated n-butanol (seeNote 7).
BIO-RAD PROTEAN-Tetra 2D comb (optional).
Razor blade, single edge no. 9.
Single lane incubation or blotting tray, such as Bio-Rad small incubation tray chamber.
Table 1.Stacking gel and separating gel solution mixture.
Stacking Gel (5 %)Separating Gel (12 %)ddH2O1.48 mL4.3 mL40 % Acrylamide solution0.25 mL3.0 mL1.0 M Tris-HCl (pH 6.8)0.25 mL-1.5 M Tris-HCl (pH 8.8)-2.5 mL10 % SDS0.02 mL0.1 mL10 % APS (seenative PAGE)0.02 mL0.1 mLTEMED (seenative PAGE)0.002 mL0.004 mLOpen in a new tab3. Methods3.1. Total Cell LysateFor native gels, cell lysis has to occur under native conditions. This can be achieved in various ways, however, some are more prone to complex dissociation or poor resolution on native gels as compared to others. One of the challenges for lysis of yeast cells is the need to disrupt the cell wall. This can be achieved enzymatically using zymolase or lyticase, however, this is not commonly used as it requires incubation at 30°C, which can induce physiological changes. More commonly, the cell wall is mechanically disrupted. In glass bead lysis, a yeast cell pellet is resuspended in lysis buffer, combined with acid-washed glass beads and vortexed for 1–2 minutes to mechanically disrupt the cell wall. Besides vortexing, specialized beading machines exist such as a bead beater. While these approaches have been used successfully, they are prone to disruption of the proteasome complexes in our hands and might require careful care to prevent warming of the sample. An alternative approach is the French Press. This lysis method has been commonly used to purify affinity-tagged proteasomes and can provide a gentle lysis. A disadvantage is that generally larger volumes of cells are required. This makes this method less convenient and more wasteful when the goal is to analyze a number of lysates on native gel (native gels require only a small amount of lysate). In our experience cryogrinding is the best approach. This can be done with liquid nitrogen using mortar and pestle. Alternatively, a grinding mill from e.g. from Retsch can be used. Here beads and jars of different volumes are available. While this approach is more reproducible and thorough as compared to grinding by hand, it is also rather cumbersome and time consuming for larger amounts of samples as it requires pair-wise processing. Our favorite approach is using small porcelain/zirconium mortars and pestles that can be connected to a cordless drill. This is done in a cryocooling box, which allows for relatively easy and reproducible creation of cryo lysates (Fig. 2).
Figure 2. Yeast total cell lysate via cryogrinding using cordless drill.
(a) Mortar and electric drill connected pestle are used for cryogrinding of small amount of cell droplets that have been frozen in liquid nitrogen. After lysis, powder can be transferred to 2 ml screwcap tube as shown and frozen for later resuspension in lysis buffer, or used directly. (b) Yeast strain with GFP-tagged β5 and mCherry-tagged Rpn1 was treated with rapamycin and cells were collected at indicated time-points, lysed by cryo-grinding and separated on native gel. Next gel was scanned on the typhoon 9410 for GFP and mCherry fluorescence (three panels on the left). Followed by an in gel suc-LLVY-AMC activity assay with 0.02% SDS. After lysis and resuspension of frozen powder in lysis buffer, 50 μl of lysate was mixed with 10 μl 6x SDS sample buffer and boiled for five minutes. 10μl was loaded on SDS-PAGE gel and immunoblotted for mCherry, GFP, and the loading control Pgk1. iCP indicates the 15S immature CP complex.
Harvest the equivalent of 100 ODs of cells (seeNote 8).
Wash cells with cold ddH2O.
Resuspend pellet in 40 μl of ddH2O.
Add liquid nitrogen to a small vessel and submerge a strainer to catch pellet. Use proper eye protection and cryo-gloves when working with liquid nitrogen and ultra-cooled materials and instruments.
Using a p200 pipette, slowly let droplets of the cell suspension drip into liquid nitrogen resulting in the formation of small frozen beads of yeast. Pellets will sink once freezing is complete. Transfer these frozen droplets to 1.5 ml tube, e.g. by using chilled tweezers (seeNote 9).
Samples can be stored at −80°C or directly used in the next step.
To lyse pellets, pour liquid nitrogen into cryocooler.
Cool mortars and pestles (one for each sample) in a small vessel with a lid (small Styrofoam box) filled with liquid nitrogen. The colder everything stays the better for grinding. We found that pre-cooling of the mortars in the cryocooler was not sufficient as it resulted in a slushy pellet.
Retrieve mortar and pestle from liquid nitrogen and place on metal rack in cryocooler (Fig. 2a).
Add the yeast frozen droplets to the mortar (For 100 ODs equivalent, we use the half of the total pellet).
Attach pestle to cordless drill and grind continuously 10 s clockwise followed by 10 s counter-clockwise until pellet becomes a fine powder. If too much pellet was added the powder can overflow the mortar here.
Scrape sides of mortar with liquid nitrogen chilled spatula (seeNote 10).
Use a tube that fits tightly on the top of the mortar to collect the material by inverting the mortar and tapping on the bottom. 2 ml screw-capped tubes fit nicely on the mortar (Fig. 2a).
Ground yeast cells can be stored at −80°C as powder, or can be resuspended in lysis buffer for further analyses.
Resuspend the ground yeast cells in proteasome lysis buffer A and clear lysates by centrifugation at 4°C at 16,000 g for 5 minutes.
Collect the supernatant. Measure protein concentration of your samples for equal protein loading. Alternatively, depending on the experimental setting, it can be preferable to load equal volumes on gel as it would represent lysates from the same amount of cells.
3.2. Purification of BaseDescribed is the method for the purification of the proteasome base complex from yeast cells harboring a protein A-tagged Rpt1 subunit. Although the base can be efficiently separated from the proteasome holoenzyme through high-salt wash (1 M NaCl) [22], the isolated base did not readily reconstitute into the proteasome holoenzyme. A major improvement in this protocol is that the base remains functional, and can be fully reconstituted into the proteasome holoenzyme [23,8]. Key to this improvement is the addition of 10% glycerol to all buffers throughout purification. Purity of the base is further improved utilizing a strain lacking RPN10 (seeNote 11). ProA-TeV-Rpt1 precipitates endogenous base in complex with the assembly chaperones, in addition to the proteasome holoenzyme. Although high-salt wash is used during base purification, some chaperones remain bound to the base due to high binding affinities between the base and chaperones [23–25]. To ensure that any endogenous chaperone is absent in the base purification, the base can be purified from yeast strains lacking individual chaperones [25]. As compared to base purification, CP purifications are more straightforward as CP is a very stable complex and does not require nucleotide unlike RP and base [22]. While these techniques purify proteasome subcomplexes, these complexes are largely derived from dissociation of assembled proteasomes holoenzyme, and thus are not true assembly intermediates. Nevertheless, they have been successfully used as model systems in reconstitution assays and various experiments to dissect the assembly process and the role of proteasome-specific assembly chaperones [25,23].
Inoculate 50 ml YPD with SY36 (a strain expressing ProA-TEV-Rpt1[22]) and grow overnight at 30°C.
Inoculate 10 ml of the overnight culture into three fernbach flasks containing 1.5 L YPD each. Grow at 30°C with shaking at 200 rpm overnight. Culture should reach an OD600 ~10 to 15.
Harvest cells by centrifugation using GS3 rotor at 4250 g for 5 min or equivalent.
Resuspend pellets in small amount of cold water and combine into one bottle. Fill bottle to top with water to wash cells (seeNote 12).
Spin down cells at 4250 g for 5 min using GS3 rotor. The wet weight of pellet will be approximately 75 grams. Store the pellet frozen at –80°C or proceed to next step.
Resuspend pellet in ~1.5 Volume of lysis buffer B. For a 75 gram pellet, use 100 ml buffer.
Lyse cells using the French Press 1250 psi, collect lysate in 250 ml centrifuge bottles. The pH of lysate should be 7 to 7.5 (seeNote 13).
To clear lysate, centrifuge lysate in GSA rotor and tubes (16,000 g for 25 min at 4°C).
Filter supernatant through cheese cloth, which will help to absorb some of the white lipid layer that typically forms on top.
Add IgG resin that has been washed in lysis buffer, to the supernatant. Use about 2.0 ml of resin for 4.5 L culture.
Incubate 1 hour at 4°C under continuous rotation.
Collect sample in BIO-RAD chromatography column (seeNote 14).
Wash with 25 bed volumes (BV) wash buffer B.
Close the column and add 5 BV dissociation buffer.
Incubate at room temperature for 1 hour under constant rotation (seeNote 15).
Wash column with 50 BV of dissociation buffer.
Wash column with 15 BV of elution buffer B.
For elution, close column and add 0.75 BV elution buffer that contains His-tagged TeV protease. Stir gently to make sure that resin and TeV protease mix.
Leave column at 30°C for 1 hour or overnight at 4°C.
Collect the first eluate.
Add ½ BV elution buffer B without TeV. Act quickly to avoid drying of resin.
Collect the second elution and repeat this step.
Combine all fractions and add 35 μl Co2+-resin to remove TeV-protease (Talon or Ni2+ -NTA resin work equally well for this purpose).
Incubate at 4°C for ~20 min under constant rotation.
Remove Co2+-resin by spinning down resin (2 min 500 g) and collecting the supernatant.
Concentrate eluate at 4°C using a centrifugal concentrator with a 10-kDa cutoff (like Centricon 10 MWCO). This procedure generally yields 100 μl of base at approximately 1 μg/μl.
3.3. Purification of ChaperonesPlasmids encoding N-terminally GST-tagged chaperones are expressed in Rosetta2 E.coli strains. Expression and purification follows standard procedures using glutathione resin. Purifications have been successful using batch approaches as well as column-based method. If necessary, chaperones can be further purified following elution by GST-tagged PreScission protease cleavage using size exclusion chromatography.
Grow Rosetta 2 cells containing plasmids of interest overnight at 37°C in LB containing chloramphenicol and antibiotics for plasmid selection (ampicillin or kanamycin).
Next day, inoculate 200 ml of LB containing relevant antibiotics with 5 ml overnight culture and grow at 37°C until OD600 = ~0.6.
Induce expression by adding IPTG to a final concentration of 0.3 mM.
Incubate cells for 3 hours at 25°C or overnight at 18°C.
Harvest cells by centrifugation. The pellet can be stored at −80°C for later processing.
Resuspend the pellet in 10 ml lysis buffer C.
Lyse by French press using manufacturers protocol
Clear lysate by centrifugation at 20,000 g for 30 min at 4°C.
Transfer supernatant to a 15 ml tube and add 200 μl Glutathione agarose resin that has been washed in lysis buffer.
Incubate for 1 hour at 4°C under constant rotation.
Spin down resin by centrifugation (500 g, 1 min, 4°C).
Wash resin with 14 ml wash buffer C.
Repeat wash four times.
Wash with 14 ml elution buffer C.
To elute chaperones and remove the GST-tag, eluate was incubated with GST-tagged PreScission, which cleaves a PreScission cleavage site located between the GST and chaperone. Here, GST-PreScission remains bound to the beads eliminating an additional step to remove the protease from the sample.
Add 1 Bed Volume elution buffer C supplemented with 5 μl PreScission Protease.
Incubate overnight at 4°C.
Precipitate resin (500 g, 1 min, 4°C) and collect supernatant.
To retrieve maximum yield, add 1 BV of elution buffer C to the beads, centrifuge and collect supernatant. Repeat twice and combine all supernatants or analyze on SDS-PAGE for fractions that contain the chaperone.
Purified chaperones can be dialyzed into reconstitution assay buffer (seesection 2.4) using a slide-a lyzer. Float slide-a-lyzer in 500 ml reconstitution assay buffer with stirring at 4°C, replace buffer after 4 hours and continue incubation overnight.
Purified chaperones can be aliquoted at stored at −80 °C.
3.4. Reconstitution Assays and Analysis of Chaperone FunctionThe proteasome holoenzyme forms via the associations of base, lid and CP. To study the function of base-binding chaperones in this process, we have established in vitro reconstitution assays. Here, we describe two strategies for using base-CP reconstitution to assay chaperone action. These strategies overcome the challenges that chaperones act rapidly and base-CP complexes are scarce in vivo. Individual chaperones can be added in specific amounts, combinations, and at specific time points during base-CP complex formation. In addition, different nucleotides (ATP, ADP, and ATPγS) can be used to create specific nucleotide-states of the base, which are linked to its maturation status during proteasome assembly [25,23]. These strategies have also been applied to examine chaperone actions during the base-lid assembly process [8,25].
3.4.1. Strategy I with free base and chaperones.Equilibrate the purified base (150–300 nM) in 10–20 μl buffer A supplemented with specific nucleotide (ADP, ATP, or ATPγS) at 1–2 mM for 10 min at room temperature. If ATP is used, include an ATP regeneration system to maintain ATP concentration during reconstitution.
To test chaperone actions, add chaperones to the base at desirable molar ratios. To ensure that the base is constantly bound by the chaperones, we add the chaperones in molar excess (for example, at 10-fold) to base. For testing the action of all three base-binding chaperones, we combine them first, and then add them simultaneously to the base (seeNote 16).
Incubate samples at room temperature for 10 min to allow for chaperone-base binding,
Add purified CP to the base at a molar ratio of CP to base of 1:2, and incubate samples at 30°C for 15, 30, and 60 min.
At the end of the incubation, briefly spin down the tubes, and add 5x native loading buffer. Immediately proceed to Native PAGE.
3.4.2. Strategy II with immobilized base and chaperonesFor preparation of resin-bound base, follow base purification procedure as specified in 3.2 up to the step immediately before TeV protease elution. Approximately 30 ml of cryo-powders generate sufficient amount of resin-bound base, which can be kept in the 4°C for several days.
Add 10 μl resin-bound base in 10–20 μl buffer A supplemented with ADP, ATP, or ATPγS at 1–2 mM. The amount of the base is estimated to be approximately 0.8 pmol. Leave the tubes for 10 min at room temperature to equilibrate the base in specific nucleotides. If ATP is used, include an ATP regeneration system to maintain ATP concentration during reconstitution.
To test chaperone actions, we add a 10-fold molar excess of chaperones to the base.
Incubate tubes at room temperature for 10 min to allow for chaperone-base binding.
Add CP at 1:2 molar ratio to the base.
Rotate the tubes in the lab rotator for 45 min in the 30°C incubator.
Collect bead-bound material by centrifugation for 15 s at 16,000 g, and wash using 500 μl of wash buffer. Repeat the washing step twice. Maintain the concentration of the nucleotides at step 2 throughout washing steps. If ATPγS was used, we typically keep it at 0.5 mM during wash since it is a non-hydrolyzable ATP analog and is also costly.
Add 50 μl of TeV elution buffer to the beads to elute base-bound complexes.
Collect the eluates by passing the material through Biorad microspin column.
Add Laemmli sample buffer and boil for 5 min at 95°C.
Analyze the samples by SDS-PAGE and immunoblotting for base subunits, chaperones, and CP (seeNote 17).
3.5. Native PAGEProteasomes and proteasome subcomplexes are well suited for separation on native PAGE and show resolutions comparable to other commonly used separating techniques, such as size exclusion chromatography or density gradient centrifugation. The disadvantage compared to these techniques, however, is that the sample will no longer be free in solution, but embedded in the gel matrix. Nevertheless, various downstream analyses and assays are still possible (Fig. 1). For native PAGE, lab-made gels with or without stacking gel are commonly used to great effect. Although costlier precast gels can be also used, but have the disadvantage that they lack ATP within the gel matrix. ATP is critical to maintain the integrity of the ATPase-containing proteasomal complexes. Samples in these native gels are resolved based on the charge, the size, and the shape of the complex. As such we have seen 26 kDa proteins migrate between complexes in the MDa range.
Prepare your mini-PAGE system by thoroughly cleaning glass plates, drying them and assembling the system according to the manufacturers instruction. We use the mini-PROTEAN tetra system from BIO-RAD, but other systems can work equally well. The native gels are very sensitive for electrophoretic anomalies. We have achieved best migration patterns for proteasomal complexes using 1.5 mm thick spacers and 10-well combs.
Prepare 10 ml of gel polymerization solution. We normally use 3.5 % to separate CP, and RP, and RP-CP complexes on gel. Acrylamide concentrations can be increased for resolving certain assembly intermediates. Take a 15 ml tube, and add 2 ml of 5X native running buffer, 0.9 ml 40% acrylamide, 7.1 ml ddH2O, and 10 μl 0.5 M ATP. Mix by inverting tube three times while avoiding bubble formation. When ready to pour gel, add 90 μl 10% APS and 9 μl TEMED, which will start the polymerization process. Mix the entire contents by inverting the tube several times (seeNote 18).
Using a serological pipette, pipet ~ 9.5 ml of the solution between the glass plates in the middle of the gel assembly using a smooth continues flow to create a homogenous gel. Stop when the top of the shorter glass plate is reached. Using a slight angle, insert the comb starting on one side. Prevent trapping of bubbles underneath the comb area to avoid electrophoretic anomalies. Close the 15 ml tube and keep on bench to monitor gel polymerization, which can take up to 30 minutes depending on temperature and light levels.
Prepare 500 ml running buffer by combining 100 ml of 5X running buffer, 500 μl ATP, and 400 ml of cold ddH2O.
Remove gel from casting stand and gently rinse wells with ddH2O. Assemble gel in electrophoresis apparatus. Fill inner chamber to the top with running buffer and use remainder for outer chamber.
Prepare samples by adding ¼ of the sample volume of 5x native loading buffer, mix by pipetting and spin solution down if needed. Carefully load samples onto the gel, using a normal p2–200 tip or gel loading tip. For the latter, be careful to avoid poking the bottom of the well. Loading empty lanes with 10 μl of 1X native sample buffer might improve overall migration behavior of neighboring lanes as well as help to track the migration of the dye. Lanes on both ends of the gel tend to generate smiling effects, similar to SDS-PAGE gels.
Add cap to the apparatus and transfer to a 4°C fridge. Run gel at ~100 Volts for 3 hours. Depending your desired migrations and specific conditions, gels can run longer or shorter. However, this period normally gives a nice separation of CP, CP-Blm10, CP-RP, and CP-RP2 from yeast when conducting in gel activity assays (seeNote 19 and Fig. 2b).
3.6. Detection of Proteasomes in Native Gels3.6.1. Fluorescent Screening of Native GelIf proteasome subunits are genetically engineered to harbor fluorescent protein, such as GFP, mCherry, CFP, or YFP, the subcomplexes containing the tagged subunit can be monitored on gels using scanning instruments with the correct lasers and filter combinations. Furthermore, fluorescent inhibitors have been developed that bind to catalytic subunits. These can be used to monitor proteasomal complexes via native gel as well [26]. We use the Typhoon 9410 scanner to detect fluorescently-tagged proteasomes in native gels. Although the gel can be scanned directly between glass plates, the plates can cause background. This can be reduced using non-luminescent glass plates (Biocraft, Japan)[10]. However, to avoid these problems, we generally disassemble the gel sandwich, and place the gel directly on the scanner plate (seeNote 20). Once gel has been placed on scanning area, use instruments settings that best match the excitation and emission spectra of the fluorescent marker. On the Typhoon, GFP can be detected using a 488 nm laser for excitation, and a 526 nm short-pass emission filter. For YFP the 532 nm laser was used with a filter 555 nm band-pass filter. To detect mCherry excitation was achieved with a 633 nm laser and data collected using a 580 nm band-pass filter. GFP and mCherry can be successfully monitored in the same gel with little cross-bleeding [27,10,28] (Fig. 2b). However, the filters are not necessarily specific to a single fluorescent protein and that some combinations result in cross-bleed of signals. Furthermore, under some settings and growing conditions a background band can be observed. Therefore, analyses should include negative control (untagged protein) and positive controls (singly tagged).
3.6.2. LLVY-AMC in Gel Activity AssayMake 20 ml of peptidase assay solution by combining 1.0 ml Tris-HCl (pH7.5) with 100 μl 1M MgCl2, 40 μl 0.5 M ATP and 100 μl 10 mM suc-LLVY-AMC and add to dish slightly bigger then mini gel, such as a square petri dish.
Once the desired electrophoresis period has been reached, remove one glass plate from the sandwich. Dislodge the gel into the assay solution carefully, as the gel tears easily under its own weight due to its low acrylamide concentration. Keep the gel facing down on the other glass plate closely above the petri dish can facilitate the transfer (seeNotes 20 and 21).
Incubate gel for 15 minutes at 30°C. For samples derived from human cells we use 37°C.
Transfer gel to your imaging system. We use a SynGENE Gbox. AMC has an optimum emission at about 460 nm and using a 440 nm band-pass filter gives us increased sensitivity as compared to standard ethidium bromide filter.
The presence of small amount of SDS (0.02%) in the assay buffer will allow for the opening of ends of the cylindrical CP, which are normally closed by the flexible N-termini of the CP α subunits. Ideally, the assays with and without SDS are done in parallel. Nevertheless, we found it very convenient to first image our samples in the absence of SDS, then add 40 μl 10% SDS per 20 ml of assay solution, incubate the gel for an additional 15 minutes and image again. The second image will show activation of free CP (Fig. 3b) as well as 26S complexes that are inhibited by Ecm29 [29,30].
Figure 3. 2D electrophoreses native-SDS-PAGE.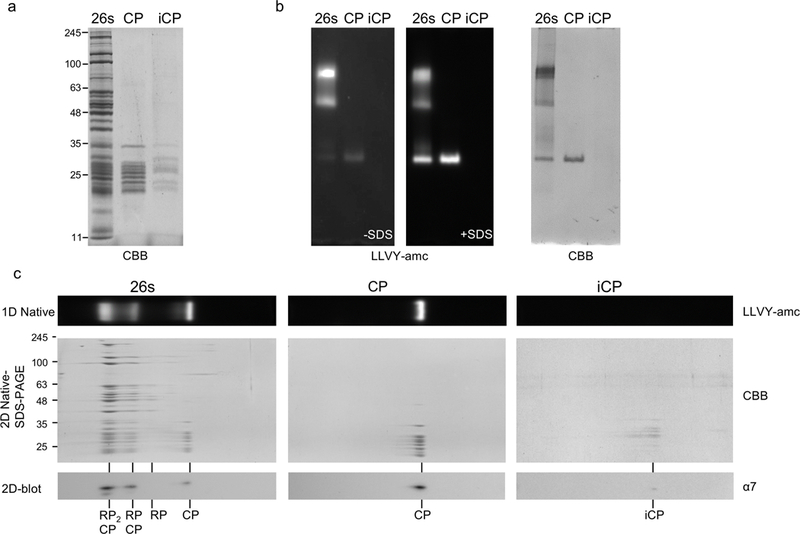
(a) Affinity purified proteasome complexes were separated on a 12% SDS-PAGE gel and stained with Coomassie dye. Protein-A tagged subunits were Rpn11, β4, and the chaperone Ump1 for 26S, CP and iCP respectively. (b) Samples were also separated on native gel, followed by suc-LLVY-AMC in gel activity assay without and with 0.02% SDS, and stained with Coomassie dyee. The SDS opens the gate of the CP allowing for entry of the substrate into free CP. RP also opens the gate thereby eliminating the need of SDS for visualization of RP-CP complexes. (c) Top panel show native gel lane excised for 2D analysis. Middle panel shows Coomassie dye stained 2D native-SDS-PAGE. Lower panel shows cropped immunoblot of 2D native-SDS-PAGE blotted for the CP subunit α7.
3.6.3. CBB Staining of GelsTransfer gel to fixing solution and incubate for 20 min on rocker.
Replace fixing solution with CBB staining solution to cover the gel (~25 ml).
Optional: microwave for ~ 10 s to speed up the staining (seeNote 22).
Incubate at room temperature while rocking until desired intensity has been reached. Transfer gel to water or destaining solution to enhance the contrast, as it will reduce gel background (seeNote 23 and Fig. 3a).
3.6.4. Immuno-blotting of Native GelsImmunoblotting of native gels can be precarious and not always straightforward. The protein complexes need to be negatively charged and bind SDS to allow transfer out of gel onto a PVDF membrane. To facilitate this, we first incubate our gel for 10 minutes in SDS-PAGE running buffer at room temperature. Next, to immunoblot the gel, standard wet transfer techniques are used (seeNote 24). Please note that the assembly of the 3.5% native gel is particularly tricky. Best approach is to first place your PVDF membrane on top of the Whatman paper, and then position the native gel on top of the PVDF, making sure to keep all wet. This allows for easier positioning as compared to assembling your native gel on the Whatman paper. Before use, the PVDF membrane has to be hydrated with methanol, followed by equilibration in ddH2O and transfer buffer. Avoid applying excessive pressure on the gel, as this could stretch or distort native gels. We generally conduct transfer at 20V (~90 mAmp) overnight in the cold room. Proceed immunoblotting following standard procedures (seeNote 24).
3.7. Two Dimensional Native-SDS PAGE.Two dimensional native to SDS-PAGE gels can provide a powerful approach to analyze samples. For purified samples, it can reveal the subunit composition of specific bands on the native gel and thus provide a more comprehensive analysis of the composition as compared to performing immunoblots for different subunits [31]. It can be also used to identify unknown factors that specifically purify with specific species of the sample. For total cell lysates, Coomassie brilliant blue or silver stain analyses are not informative due to the large amount of protein complexes, therefore we combine 2D gels with immunoblotting. While native gels can be directly used for subsequent immunoblotting, this can be challenging under some circumstances. The extra step of a second dimension prior to immunoblotting has two major advantages. First, detection sensitivity for the protein of interest increase substantially. This suggests that the SDS-PAGE step either results in better denaturing or exposure of epitopes of the proteins, or the transfer efficiency is enhanced for migration from SDS-PAGE to PVDF as compared to native page to PVDF. Second, it provides molecular weight information. This can be valuable if the protein of interest is modified post translationally. Furthermore, antibodies might show cross-reactivity with other proteins of a different molecular weight. Since in native-PAGE polypeptides migrate in complexes, the molecular weight information is absent. Thus, careful controls should be included and caution should be taken in interpretation of immunoblot signals of native-PAGE. Including a second dimension of SDS-PAGE in analyses provides the molecular weight information, thereby allowing for a more reliable interpretation of the immunoblot data (Fig. 3c).
Make 12% minigel according to manufacturer’s instruction. After polymerization of the separating gel, rinse minigel with ddH2O. Next, prepare and pour the stacking gel using either a 2D gel comb if available or add stacking solution till a little below the top of the short plate (seeNote 25). For the latter, use water saturated n-butanol to create a straight surface that is sealing the air for efficient polymerization. Once ready to proceed with 2D gel, remove comb from the SDS-PAGE gel and rinse with ddH2O.
Excise lane (or bands) of interest from your native gel (seeNote 26). Carefully transfer the gel piece to e.g. a Bio-Rad small incubation tray chamber filled with 1x SDS-PAGE sample buffer. Incubate under gentle rocking for 15 minutes (seeNote 27).
Prepare an agarose solution by dissolving 0.5% agarose in 1x SDS running buffer (seeNote 28). Once fully dissolved (look carefully for the absence of any transparent clumps) transfer the solution to a couple of 1.5 ml tubes and keep in heating block > 65°C to prevent premature solidification. This solution is used to seal the native gel piece in place on top of the second dimension SDS PAGE gel (seestep 6 below).
Carefully take your native gel lane from the 1X SDS sample buffer and position horizontal on the separating gel. Carefully slide/push the gel in position (see Note 29).
Secure the gel lane into position by pipetting melted agarose solution on and around the gel piece (seeNote 30).
Once solidified, assemble the SDS-PAGE gel into the running apparatus, fill chambers with running buffer, load protein marker and run gel as you would run a normal SDS-PAGE gel. Then proceed to further downstream analyses like immunoblotting or CBB or silver staining.
4. NotesBoth ends of the CP can bind these regulators and hybrid forms exists, e.g. where Blm10 binds to one end of the CP and RP the other end (Blm10-CP-RP). As such, even purifications with a tag to one of the regulators can yield other regulators in a purification.
Another convenient indicator of assembly intermediates exists for the CP, since five β subunits (β1, β2, β5, β6, β7) contain a propeptide that are proteolytically removed upon formation of mature CP. The size of these subunits is thus a reliable indicator of the assembly stage.
This solution is made by combining 54.5 g Tris-Base, 27.8 g Boric Acid, 25 ml 1 M MgCl2, and 5 ml 0.5 M EDTA (pH 7.5), pH is not adjusted and this combination of chemicals should result in a pH a bit higher than 8. The solution is filter sterilized, stored at 4° C, and can be used up to a few months.
Solution is stable at 4°C for ~ 2–3 weeks, or aliquots can be stored in −20°C for longer periods avoid repeated freeze-thaw cycles.
The suc-LLVY-AMC (LLVY stands for the amino acids that are part of this peptide) is light sensitive and the stock should be kept in dark. During assay period no special protection from light is generally required. Over time this solution has a tendency to precipitate. If the solution precipitates, the tube can be centrifuged to precipitate the crystalized material. Normally, sufficient amount of substrate remains in solution and in gel activity assays are not impeded. Using the solution in more quantitative activity assays is not advised. An alternative solvent is DMSO.
suc-LLVY-AMC is most commonly used and monitors mainly the chymotrypsin-like active sites (β5 residues). However, substrates are commercially available to monitor trypsin like (β2) or caspase-like (β1) activity if desired [32].
Water-saturated butanol prevents drying of the gel by absorption of water from the butanol solution. Mix water and butanol thoroughly, let phases separate and used is top layer. Due to the higher difference in density as compared to simply using water to layer on top of the polymerizing solution, the butanol provides a straight and sharp edge. Rinse away butanol using ddH2O prior to proceeding to next step.
Culture size depends on the physiological state of the sample you desire. Normally, logarithmically growing cells are desired. To achieve this, we generally inoculate a 100 ml culture to OD600 ~ 0.2 using a small overnight culture. These cells are grown for several hours till OD ~1 is reached, spinning down the cells in this 100 ml culture will give you the 100 OD equivalent of cells.
Do not yet seal the lid as it will pop open from evaporating N2 (l) and some pellet can be lost.
We found one half of a flat-ended tweezer with the far end wrapped in tape works well to scrape out the material that remains in the bottom of the mortar.
The purification of base from Rpt1-tagged strains yields a small amount of 26S proteasomes that are resistant to treatment aimed at proteasome dissociation. The use of an Rpt1-tagged strain that has a deletion of RPN10 eliminates the residual 26S proteasome in the base purification.
Omitting wash step can lower the pH of the lysate and cause premature dissociation of proteasome complexes.
Alternatively, yeast cells can be directly frozen as droplets and ground under cryogenic conditions using a Retsch Mill.
Use appropriate column for resin volume; for 0.75 ml resin, we use the 1.5 cm diameter, which actually gives a rather small resin bed. For 2 ml resin, a bigger column can be used.
This step leads to the disruption RP into the lid and base. We close top and bottom of column and attach it horizontal to a lab-quake and keep it rotating. Just leaving the column at room temperature without movement does not appear to give good disruption. Also, we observed better dissociation at RT as compared to 4°C.
To test chaperone actions on pre-formed base-CP complexes, allow the base-CP complexes to form first, and then add the specific chaperones of interest.
Instead of using immobilized base, immobilized CP can be used [23] for base-CP reconstitution. In this case, free base and chaperones are added at specific points during the assays.
Native PAGE can be prone to leakage. This can be reduced by avoiding chipped glass plates, careful attention to lining up the bottoms of glass plates and spacers. Most leakage occurs at the ends where glass plates and spacers meet, using a very small amount of Vaseline only here can greatly reduce the occurrence of leaks.
Note that the dye reaches the bottom of the gel after about 2 hours, but longer running times are often preferred. If gels are scanned for fluorescence, they can still be assayed for in gel activity followed by CBB staining or immunoblotting.
To facilitate the handling of these low percentage native gels that have a consistency weaker than stacking gels, keeping surfaces wet is crucial. Also, gels should not be pinched between two fingers, as the weight of the hanging gel will cause tearing. Instead, roll up the gel during handling, grab it all at once with all five fingers, or fold it once and grab it as a whole using all five fingers.
To easily dislodge the gel from the plate the green wedge that BIO-RAD provides to separate the glass plates can be used to run it through the corners to detach the gel from the edges of glass plate.
Solution should not boil during this step, if so shorten time in microwave.
CBB R-250 is less suited for this protocol. Both CBB chemicals have similar sensitivity, however, CBB R-250 will cause strong staining of gel and bands are less prominent after de-staining.
The transfer to PVDF can be done with or without 20% methanol. We routinely eliminate methanol to reduce organic waste, however, for some proteins the methanol can be crucial. For example, the presence of methanol in the transfer dramatically increases the detection of free ubiquitin.
The percentage of the SDS-PAGE can be adjusted based on the MW of the proteins of interest. Our standard 11 or 12% allows a reasonable separation of the lower molecular weight CP subunits and provides a good separation of the higher molecular weight proteasome subunits without the need for a gradient gel.
Doing the excision of the lane on a UV box after the activity assay will facilitate this process as the activity in the different lanes can be used to guide the excision. Avoid back-forward saw/cutting motion. We normally push a razorblade straight down to cut an area, raise the blade, and move to the next part of the region to be cut. Others have used a pizza cutter [33].
Whenever handling a lane form native gel, holding it on one edge can cause the gel to rip under its own weight. Carefully position e.g. a wide razor blade or the green plate wedge from BIO-RAD underneath the center of the gel strip to lift the gel.
We generally use a 50 ml tube filled with 10 ml of 1x SDS running buffer and ~ 50 mg of agarose. Heat in microwave in short bursts to avoid over boiling with the SDS.
Align the excised lane on top of separating gel. Here, we generally position top of the native gel strip on the side where we load the marker. Be careful to prevent trapped bubbles or gaps, as they will cause deformations when running the second dimension. Use gentle pressure to push the gel strip into position and partly between the two glass plates starting from one end. A blunt thin object, like the opposite end of the razor blade, will facilitate this process. Deformations from a straight angle of the strip with respect to the separating gel results in broadening of the dots on the final 2D gel in the x-axis direction.
2D-gel combs will have a lane for protein marker. If not using 2D comb, prior to adding agar, insert a single 1.5 mm thick comb tooth next to the gel lane in the agarose for loading of the protein marker.
AcknowledgementsMany of these techniques described above have been developed or optimized and tweaked by many experts in the field and the power of their use has been displayed in numerous excellent papers. We apologize for not being able to provide a comprehensive reference to all those contributions in this publication. This work was supported in part by grants to J.R. from NIH (R01-GM118660 and R15-GM112142).
References1.Kressler D, Hurt E, Bassler J (2017) A Puzzle of Life: Crafting Ribosomal Subunits. Trends Biochem Sci42 (8):640–654. doi: 10.1016/j.tibs.2017.05.005 [DOI] [PubMed] [Google Scholar]2.Ellis RJ (2013) Assembly chaperones: a perspective. Philos Trans R Soc Lond B Biol Sci368 (1617):20110398. doi: 10.1098/rstb.2011.0398 [DOI] [PMC free article] [PubMed] [Google Scholar]3.Budenholzer L, Cheng CL, Li Y, Hochstrasser M (2017) Proteasome Structure and Assembly. J Mol Biol. doi: 10.1016/j.jmb.2017.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]4.Wani PS, Rowland MA, Ondracek A, Deeds EJ, Roelofs J (2015) Maturation of the proteasome core particle induces an affinity switch that controls regulatory particle association. Nat Commun6:6384. doi: 10.1038/ncomms7384 [DOI] [PMC free article] [PubMed] [Google Scholar]5.Ramos PC, Hockendorff J, Johnson ES, Varshavsky A, Dohmen RJ (1998) Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell92 (4):489–499. doi:S0092-8674(00)80942-3 [DOI] [PubMed] [Google Scholar]6.Kock M, Nunes MM, Hemann M, Kube S, Dohmen RJ, Herzog F, Ramos PC, Wendler P (2015) Proteasome assembly from 15S precursors involves major conformational changes and recycling of the Pba1-Pba2 chaperone. Nat Commun6:6123. doi: 10.1038/ncomms7123 [DOI] [PubMed] [Google Scholar]7.Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Shi Y, Zhang N, de Poot SA, Tuebing F, Sun S, Vannoy J, Tarasov SG, Engen JR, Finley D, Walters KJ (2016) Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science351 (6275). doi: 10.1126/science.aad9421 [DOI] [PMC free article] [PubMed] [Google Scholar]8.Park S, Roelofs J, Kim W, Robert J, Schmidt M, Gygi SP, Finley D (2009) Hexameric assembly of the proteasomal ATPases is templated through their C termini. Nature459 (7248):866–870. doi: 10.1038/nature08065 [DOI] [PMC free article] [PubMed] [Google Scholar]9.Roelofs J, Park S, Haas W, Tian G, McAllister FE, Huo Y, Lee BH, Zhang F, Shi Y, Gygi SP, Finley D (2009) Chaperone-mediated pathway of proteasome regulatory particle assembly. Nature459 (7248):861–865. doi: 10.1038/nature08063 [DOI] [PMC free article] [PubMed] [Google Scholar]10.Saeki Y, Toh EA, Kudo T, Kawamura H, Tanaka K (2009) Multiple proteasome-interacting proteins assist the assembly of the yeast 19S regulatory particle. Cell137 (5):900–913. doi: 10.1016/j.cell.2009.05.005 [DOI] [PubMed] [Google Scholar]11.Funakoshi M, Tomko RJ Jr., Kobayashi H, Hochstrasser M (2009) Multiple assembly chaperones govern biogenesis of the proteasome regulatory particle base. Cell137 (5):887–899. doi: 10.1016/j.cell.2009.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]12.Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J (2010) Assembly, structure, and function of the 26S proteasome. Trends Cell Biol20 (7):391–401. doi: 10.1016/j.tcb.2010.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]13.Tomko RJ Jr., Hochstrasser M (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem82:415–445. doi: 10.1146/annurev-biochem-060410-150257 [DOI] [PMC free article] [PubMed] [Google Scholar]14.Higashitsuji H, Liu Y, Mayer RJ, Fujita J (2005) The oncoprotein gankyrin negatively regulates both p53 and RB by enhancing proteasomal degradation. Cell Cycle4 (10):1335–1337. doi: 10.4161/cc.4.10.2107 [DOI] [PubMed] [Google Scholar]15.Tomko RJ Jr., Funakoshi M, Schneider K, Wang J, Hochstrasser M (2010) Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol Cell38 (3):393–403. doi: 10.1016/j.molcel.2010.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]16.Kusmierczyk AR, Kunjappu MJ, Funakoshi M, Hochstrasser M (2008) A multimeric assembly factor controls the formation of alternative 20S proteasomes. Nat Struct Mol Biol15 (3):237–244. doi: 10.1038/nsmb.1389 [DOI] [PubMed] [Google Scholar]17.Tomko RJ Jr., Taylor DW, Chen ZA, Wang HW, Rappsilber J, Hochstrasser M (2015) A Single alpha Helix Drives Extensive Remodeling of the Proteasome Lid and Completion of Regulatory Particle Assembly. Cell163 (2):432–444. doi: 10.1016/j.cell.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]18.Estrin E, Lopez-Blanco JR, Chacon P, Martin A (2013) Formation of an intricate helical bundle dictates the assembly of the 26S proteasome lid. Structure21 (9):1624–1635. doi: 10.1016/j.str.2013.06.023 [DOI] [PubMed] [Google Scholar]19.Kaneko T, Murata S (2012) Using siRNA techniques to dissect proteasome assembly pathways in mammalian cells. Methods Mol Biol832:433–442. doi: 10.1007/978-1-61779-474-2_30 [DOI] [PubMed] [Google Scholar]20.Tallec BL, Peyroche A (2012) Using DNA damage sensitivity phenotypes to characterize mutations affecting proteasome function. Methods Mol Biol832:363–371. doi: 10.1007/978-1-61779-474-2_25 [DOI] [PubMed] [Google Scholar]21.Barrault MB, Richet N, Godard C, Murciano B, Le Tallec B, Rousseau E, Legrand P, Charbonnier JB, Le Du MH, Guerois R, Ochsenbein F, Peyroche A (2012) Dual functions of the Hsm3 protein in chaperoning and scaffolding regulatory particle subunits during the proteasome assembly. Proc Natl Acad Sci U S A109 (17):E1001–1010. doi: 10.1073/pnas.1116538109 [DOI] [PMC free article] [PubMed] [Google Scholar]22.Leggett DS, Glickman MH, Finley D (2005) Purification of proteasomes, proteasome subcomplexes, and proteasome-associated proteins from budding yeast. Methods Mol Biol301:57–70. doi: 10.1385/1-59259-895-1:057 [DOI] [PubMed] [Google Scholar]23.Park S, Li X, Kim HM, Singh CR, Tian G, Hoyt MA, Lovell S, Battaile KP, Zolkiewski M, Coffino P, Roelofs J, Cheng Y, Finley D (2013) Reconfiguration of the proteasome during chaperone-mediated assembly. Nature497 (7450):512–516. doi: 10.1038/nature12123 [DOI] [PMC free article] [PubMed] [Google Scholar]24.Le Tallec B, Barrault MB, Guerois R, Carre T, Peyroche A (2009) Hsm3/S5b participates in the assembly pathway of the 19S regulatory particle of the proteasome. Mol Cell33 (3):389–399. doi: 10.1016/j.molcel.2009.01.010 [DOI] [PubMed] [Google Scholar]25.Li F, Tian G, Langager D, Sokolova V, Finley D, Park S (2017) Nucleotide-dependent switch in proteasome assembly mediated by the Nas6 chaperone. Proc Natl Acad Sci U S A114 (7):1548–1553. doi: 10.1073/pnas.1612922114 [DOI] [PMC free article] [PubMed] [Google Scholar]26.Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, van der Linden WA, van den Nieuwendijk AM, Hofmann T, Berkers CR, van Leeuwen FW, Groothuis TA, Leeuwenburgh MA, Ovaa H, Neefjes JJ, Filippov DV, van der Marel GA, Dantuma NP, Overkleeft HS (2006) A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol13 (11):1217–1226. doi: 10.1016/j.chembiol.2006.09.013 [DOI] [PubMed] [Google Scholar]27.Waite KA, De-La Mota-Peynado A, Vontz G, Roelofs J (2016) Starvation Induces Proteasome Autophagy with Different Pathways for Core and Regulatory Particles. J Biol Chem291 (7):3239–3253. doi: 10.1074/jbc.M115.699124 [DOI] [PMC free article] [PubMed] [Google Scholar]28.Enenkel C (2012) Using native gel electrophoresis and phosphofluoroimaging to analyze GFP-tagged proteasomes. Methods Mol Biol832:339–348. doi: 10.1007/978-1-61779-474-2_23 [DOI] [PubMed] [Google Scholar]29.Lee SY, De la Mota-Peynado A, Roelofs J (2011) Loss of Rpt5 protein interactions with the core particle and Nas2 protein causes the formation of faulty proteasomes that are inhibited by Ecm29 protein. J Biol Chem286 (42):36641–36651. doi: 10.1074/jbc.M111.280875 [DOI] [PMC free article] [PubMed] [Google Scholar]30.De La Mota-Peynado A, Lee SY, Pierce BM, Wani P, Singh CR, Roelofs J (2013) The Proteasome-associated Protein Ecm29 Inhibits Proteasomal ATPase Activity and in Vivo Protein Degradation by the Proteasome. J Biol Chem288 (41):29467–29481. doi: 10.1074/jbc.M113.491662 [DOI] [PMC free article] [PubMed] [Google Scholar]31.Kleijnen MF, Roelofs J, Park S, Hathaway NA, Glickman M, King RW, Finley D (2007) Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat Struct Mol Biol14 (12):1180–1188. doi: 10.1038/nsmb1335 [DOI] [PubMed] [Google Scholar]32.Li Y, Tomko RJ Jr., Hochstrasser M (2015) Proteasomes: Isolation and Activity Assays. Curr Protoc Cell Biol67:3 4341–20. doi: 10.1002/0471143030.cb0343s67 [DOI] [PMC free article] [PubMed] [Google Scholar]33.Hochstrasser M, Funakoshi M (2012) Disulfide engineering to map subunit interactions in the proteasome and other macromolecular complexes. Methods Mol Biol832:349–362. doi: 10.1007/978-1-61779-474-2_24 [DOI] [PMC free article] [PubMed] [Google Scholar]